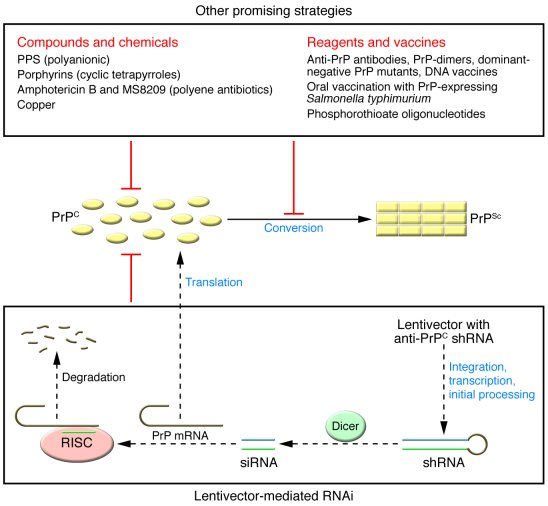
Prion disease treatment is an emerging field that holds promise for combating some of the most devastating neurodegenerative disorders known to science. These rare conditions, including Creutzfeldt-Jakob disease and fatal familial insomnia, are caused by misfolded prion proteins that inflict irreversible damage to the brain. Recent advancements in gene editing therapy have sparked hope among researchers and patients alike, providing a potential pathway toward alleviating the impacts of these fatal diseases. Although the journey to clinical application remains complex and filled with scientific challenges, the results from groundbreaking studies are paving the way for innovative therapies. As investigations into prion diseases unfold, the commitment of researchers like Sonia Vallabh and Eric Minikel highlights the personal stakes involved in finding effective treatments.
The treatment for prion diseases is a critical area of research aimed at addressing the grave challenges posed by these lethal neurodegenerative conditions. Characterized by protein misfolding, disorders such as Creutzfeldt-Jakob disease and fatal familial insomnia are notorious for their rapid progression and lethal outcomes. Innovative gene-editing strategies, aimed at correcting the mutations responsible for these diseases, represent a significant leap forward in potential therapeutic options. As researchers harness advanced techniques to modify genetic structures, the prospect of transforming a fatal diagnosis into a manageable condition becomes increasingly real. This multidisciplinary approach underscores the importance of collaborative efforts among scientists and the personal motivation driving this vital work.
Understanding Prion Diseases
Prion diseases represent a group of rare but devastating neurodegenerative disorders characterized by the misfolding of prion proteins in the brain. Among the well-known types of prion diseases are Creutzfeldt-Jakob disease, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome. These conditions lead to progressive brain damage and are invariably fatal, with symptoms including severe cognitive decline and loss of motor function. According to recent research, about 15% of prion disease cases stem from inherited mutations in the prion protein gene, while the remaining 85% occur sporadically without a clear genetic cause.
The misfolding of prion proteins has serious implications for brain health, as these abnormal proteins can induce other normally folded proteins to misfold as well, creating a dangerous cascade. This attribute of prion diseases, termed “infectious protein misfolding,” contributes to their classification not only as genetic but also as transmissible diseases, causing alarm in both the medical and scientific communities.
Furthermore, prion diseases often present unique challenges in diagnosis and treatment due to their asymptomatic early stages and the rapid progression once symptoms appear. In much of the medical literature, prion diseases are linked to other neurodegenerative disorders, underlining the urgency for effective therapies. Despite significant advancements in understanding the biology of these diseases, the pressing need for novel and effective treatment options is evident, particularly as the aging population increases the prevalence of neurodegenerative conditions overall.
Breakthroughs in Prion Disease Treatment
Recent advancements in gene editing therapy present a beacon of hope for treating prion diseases like Creutzfeldt-Jakob disease and fatal familial insomnia. Research conducted by scientists at the Broad Institute has demonstrated the capability of a groundbreaking gene-editing technique known as base editing. This approach allows for precise alterations to the DNA coding for the misfolded prion proteins, significantly reducing their levels in animal models. In lab mice, this treatment decreased the presence of harmful prion proteins by nearly 50%, resulting in a remarkable extension of life expectancy, highlighting the potential of such therapies in the future clinical treatment of humans suffering from these disorders.
As the data suggest, this method not only offers a glimpse into viable treatment pathways but also enhances the understanding of the underlying mechanisms of prion diseases. However, experts emphasize caution, as several hurdles must be addressed before these promising findings can translate into treatments for human patients. Ensuring the safety and efficacy of these therapies will require extensive research and multiple clinical milestones to be overcome.
The collaborative efforts among researchers, including patient-scientists Sonia Vallabh and Eric Minikel, have propelled the study of prion diseases into promising new territories. Their personal connection to fatal familial insomnia adds a layer of urgency and motivation to their work, producing meaningful advancements in contemporary neuroscience. Current research initiatives have identified key strategies to optimize gene editing techniques, modifying vector delivery systems to improve targeting and minimize unintended consequences, which are crucial for future human applications.
The Role of Patient-Scientists in Research
The engagement of patient-scientists in prion disease research highlights a transformative approach where personal experience drives scientific inquiry and innovation. Sonia Vallabh and Eric Minikel, both deeply affected by familial prion disease, embody this model, transforming their grief and concern into a concerted effort to understand and combat these neurodegenerative disorders. Their involvement not only enriches the research with unique insights but also fosters a sense of urgency and purpose among their colleagues, ultimately enhancing collaborative efforts within the scientific community.
The patient-scientist model emphasizes the significance of personal narratives in the scientific process. These individual stories can inspire researchers and resonate deeply within the broader community, fostering support for scientific endeavors aimed at curing fatal conditions like prion diseases. By bridging the gap between clinical experience and laboratory research, patient-scientists are proving instrumental in pushing the boundaries of traditional research paradigms, leading to more comprehensive and impactful approaches toward the treatment of complex neurodegenerative diseases.
Future Directions for Prion Disease Therapies
As research on prion diseases progresses, the outlook for clinical therapies seems increasingly optimistic. The foundational work being conducted in laboratories is setting the stage for subsequent phases that will involve rigorous human trials. Researchers are actively addressing critical questions related to the safety of gene editing interventions and how they can be effectively administered to human patients. This inquiry is particularly pertinent as prion diseases are marked by their unique and potentially hazardous progression, necessitating tailored therapeutic strategies that consider both genetic and environmental factors.
Future efforts are likely to focus on refining gene editing technologies to enhance their efficiency and safety. Innovations in vector systems for gene delivery, as well as improved methodologies for targeting specific tissues in the brain, will be crucial. Collaborative initiatives, such as those emerging from institutions like the Broad Institute, are expected to spearhead these advancements, facilitating a more translational approach where laboratory discoveries translate swiftly into clinical applications.
Preventing Prion Disease Transmission
The transmission of prion diseases is a significant area of concern, particularly in medical and research settings. The alarming contagious nature of prions underscores the importance of stringent control measures to prevent their dissemination. Understanding the pathways through which prions spread and identifying potential sources of infection are critical for safeguarding individuals and the public health infrastructure. This includes rigorous sterilization protocols for medical instruments and biohazard containment in research environments where prion exposure might occur.
More widely, public health policies must adapt to incorporate knowledge about prion diseases, ensuring that healthcare professionals are educated on proper prevention methods. This awareness is particularly vital in countries where livestock is affected by prion diseases like Bovine Spongiform Encephalopathy (BSE), which can have serious implications for both animal and human health. Future public health strategies must focus on reducing the risk of incidents involving accidental exposure and educating communities about the realities of prion disease transmission.
Potential of Gene Editing in Neurodegenerative Research
The integration of gene editing therapy into the study of neurodegenerative diseases represents a paradigm shift in scientific research and treatment methodologies. Advances in techniques such as CRISPR and base editing are unlocking new possibilities for targeting the genetic foundations of various conditions, including prion diseases. This intersection of genetic manipulation and therapeutic intent underscores the potential for innovative solutions to combat disorders characterized by progressive neural degeneration, paving the way for future treatments that could ultimately alter the trajectory of diseases like Creutzfeldt-Jakob disease.
Moreover, the potential for gene editing to target the underlying causes of neurodegenerative disorders extends the horizons of what is achievable in neurological research. As methodologies continue to evolve, so too will the understanding of diseases at both the molecular and systemic levels. The hope is that such progress will not only open doors for treating prion diseases but will also provide insights applicable to other neurodegenerative conditions, creating a comprehensive arsenal against a myriad of challenges facing scientists and clinicians alike.
The Impact of Research Collaboration
Collaboration among researchers is essential for driving advancements in the understanding and treatment of complex diseases such as prion disorders. Interdisciplinary partnerships enable the pooling of diverse expertise and resources, fostering an environment ripe for innovation. This collaborative spirit is evident in the joint efforts at institutions like the Broad Institute, where experts from various fields converge to tackle the elusive nature of prion diseases and explore potential therapeutic avenues through gene editing.
The involvement of multiple research teams contributes to a more robust scientific approach, increasing the likelihood of breakthroughs that can ultimately lead to clinical applications. Such collaborations not only enhance the quality of research but also create avenues for knowledge exchange and mentorship, vital for nurturing the next generation of scientists dedicated to combating neurodegenerative diseases.
Challenges in Developing Prion Disease Therapies
Despite the promise shown by recent studies in the treatment of prion diseases, significant challenges remain in translating laboratory successes into effective therapies for human patients. One major hurdle is ensuring safety in gene editing approaches, particularly given the innate complexities of the central nervous system and the risk of off-target effects. These challenges necessitate extensive preclinical testing and careful evaluation of potential side effects to ensure the well-being of future patients.
Moreover, researchers must navigate regulatory landscapes that complicate the speed at which new treatments can be brought to market. Ensuring compliance with ethical standards, especially in human trials involving experimental therapies, is crucial for maintaining public trust in scientific progress. Those involved in prion disease research must advocate for a balanced approach that prioritizes rigorous scientific inquiry while expediting the path to viable treatments.
Patient Advocacy and Awareness
Increasing awareness around prion diseases is a critical aspect of advancing research and treatment options. Patient advocates play a powerful role by sharing their experiences and educating the public and healthcare professionals about the realities of these disorders. This grassroots effort not only enhances understanding but also helps to alleviate the stigma associated with neurodegenerative diseases, fostering support for research initiatives aimed at finding cures.
Moreover, patient advocacy efforts can empower affected families to seek out and participate in clinical trials, thereby accelerating the research process. Engaging communities and stakeholders creates a network of support that enhances the visibility of prion diseases and propels funding opportunities, ultimately fueling progress in therapeutic development. By amplifying the voices of those directly impacted by these conditions, advocacy groups help bridge the gap between patient needs and scientific inquiry.
Frequently Asked Questions
What advancements have been made in prion disease treatment using gene editing therapy?
Recent research has shown promising advancements in prion disease treatment through innovative gene editing therapy. A study from the Broad Institute demonstrated that altering a single base in the gene responsible for producing harmful prion proteins can significantly reduce their levels in the brain, potentially leading to effective treatments not just for Creutzfeldt-Jakob disease but also for related neurodegenerative disorders.
How does gene editing therapy target prion diseases like fatal familial insomnia?
Gene editing therapy specifically targets prion diseases, including fatal familial insomnia, by modifying the gene that encodes for the prion protein. Researchers have successfully tested this technique in mice, showing a reduction in the amount of toxic prion proteins, which could pave the way for future treatments in humans affected by hereditary forms of prion disease.
Are there potential clinical trials for treating Creutzfeldt-Jakob disease with gene editing?
While promising results have emerged from gene editing techniques, potential clinical trials for treating Creutzfeldt-Jakob disease with these therapies are still several years away. Research continues to ensure the safety and efficacy of these methods before they can be tested in human participants.
What role do patient-scientists play in developing treatments for prion diseases?
Patient-scientists, such as Sonia Vallabh and Eric Minikel, play a crucial role in the development of treatments for prion diseases. Their personal experiences with fatal familial insomnia drive their research and collaboration with other experts, fostering a unique motivation to advance gene editing therapies for prion-related disorders.
How does the research on prion disease treatment align with advancements in neurodegenerative disorders?
The research focused on prion disease treatment is significantly aligned with advancements in the study of neurodegenerative disorders. Insights gained from gene editing therapy applied to prion diseases may also inform strategies for other neurodegenerative conditions, as many share similar pathways related to protein misfolding.
What are the challenges faced in developing a treatment for prion disease?
Developing a treatment for prion disease presents several challenges, including ensuring the safety of gene editing techniques and overcoming the complexity of delivering the therapy effectively. Researchers must refine the base editing technology and conduct extensive preclinical testing before initiating human trials.
What is the significance of the recent findings in addressing inherited prion diseases?
The recent findings concerning gene editing therapy are significant as they offer a potential strategy for addressing inherited prion diseases like fatal familial insomnia. By specifically targeting the genetic mutations responsible for protein misfolding, there is hope for meaningful therapeutic interventions in the near future.
| Key Points | Details |
|---|---|
| Research Milestone | Recent advances in gene-editing therapy show potential for treating prion diseases. |
| Ethical Motivation | Researchers, including Sonia Vallabh, are personally affected by prion diseases, fostering a deep commitment to their work. |
| Mouse Model Success | Gene-editing reduced toxic prion protein by half and extended mouse lifespan by 52%. |
| Collaboration Importance | Collaboration among experts enhances the potential for breakthroughs in treatments. |
| Future Steps Needed | Further research and safety refinements are needed before human trials can commence. |
Summary
Prion disease treatment is becoming a more tangible goal thanks to recent advancements in gene-editing technologies. Groundbreaking research found that altering a single base in a gene associated with fatal prion disorders significantly reduced toxic protein levels in laboratory mice while extending their lifespans. The personal connection of the researchers to prion diseases fuels their motivation, creating an effective collaboration aimed at developing viable therapies. Although human trials remain several years away, the promising results pave the way for future innovations in tackling these devastating conditions.